PAINT User Guide
Summary
PAINT is a Java application for viewing and annotating phylogenetic trees. The present document describes how to set up and use the tool.
Requirements
Java 1.8 (aka Java 8 on a Macintosh) must be installed.
Installing and configuring PAINT
PAINT is a Java application, and can be run on a either Mac or Windows. To install PAINT, download the application from: http://paintcuration.usc.edu/
Launching PAINT
- On a Windows machine, run the program
lauchPAINT.bat. - On a Mac, open a Unix terminal window, go to the directory containing the PAINT program, and execute the command:
sh launchPAINT.sh OR ./launchPAINT.sh
Using PAINT
Login
You are required to login before you can open a tree. The purpose is to record proper acknowledgement for all the curated annotations (of tree nodes) created by you.
Go to File -> Login.
If you just want to view the tree and annotations, you can enter gouser as the username. The password is filled already. This is a read-only login.
If you want to curate trees, enter your username and password. If you don’t have a login and password, send an email to huaiyumi@usc.edu and request one.
Curating a gene family
The analogy is to a library. You will first find and check out (lock) the families you want to curate, and then select a family to curate from your list of locked families. All families now have a curation status (curated, partially curated, uncurated).
Step 1: Find and "lock" families for curation
When you lock the family, other curators won’t be able to curate them. This is to prevent people from working on the same family.

- Go to
File>Manage and View Books...- A window will pop up (Fig 1).
- You can search for families by various identifiers: PTHR ID, PTN ID, Gene Symbol, Protein Identifier, Gene Identifier, or gene definition.
- Retrieve a list of all families, or just the uncurated families.
- Retrieve a list of families that require review (incompletely curated).
- Press the "submit" button to launch search
- A window will pop up (Fig 1).

- Select one or several families to lock. Fig 2 shows an example when all uncurated families are returned. There are 4 possible curation status states:
- Manually curated – These are the families curated, and the curator believes that the curation is complete.
- Locked – Those families are locked by a curator. The name of the curator who locks the family is shown in the Locked by column.
- Partially curated – These are the families that have been curated. The curator can unlock the family and leave it as partially curated.
- Require PAINT review – The previously curated paint annotations are changed due to updates in either PANTHER and GO.
- Unknown – These are uncurated families.
- Check the box in the
Lock/Unlockcolumn of the families you want to check out, and clickLock or Unlock selected Booksbutton at the bottom of the panel.
Step 2: Open a family to curate
- To open a family, click
View Locked Books, and then click theViewbutton (Fig 3). - A family can only be locked by a single user. If a family is already locked, you can open it in View Only mode.
- You can only curate families you have locked.

Step 3: Save your annotations
You can choose to save but keep the family locked so you can continue the curation later. You can also save and unlock the family.
- Go to
File>Save to Database. A window will pop up with the following options:- Cancel
- Save and unlock: The family will be unlocked and marked as Partially Curated.
- Save: The family will remain locked. The curator should do this as often as possible during the curation.
- Save, unlock & set curated: The family will be marked as Manually Curated.
Appearance and Basic Operation
Windows
PAINT is organized into three main panels (Fig 4):

- The upper left panel shows a phylogenetic tree.
- The upper right panel allows you to switch back and forth between (i) the Annotation Matrix; (ii) the Protein Information Table and (iii) a multiple sequence alignment (MSA) of all sequences.
- The bottom panel contains two tabs: Annotations and Evidence.
All the tabbed panes may be resized or split out into windows.
- Click on a tab (e.g., Protein Information, Evidence) to bring it to the front.
- Click the icons in the tabs or the upper right corner to Undock/Dock, Minimize, Maximize, or close individual tabs or groups of tabs.
- Tabs and panes may also be rearranged within a window by dragging.
- Columns in the Protein Information Table can be resized.
- Windows may be closed, arranged, or resized by dragging boundaries.
Recommended configuration for curation
- Bigger is better. Use as much of the monitor as you can afford. If you are using a laptop, you may wish to attach an external monitor.
- Adjust the width of the window and the partition between the Tree and the Table until you are comfortable with them.
Phylogenetic Tree
A phylogenetic tree contains nodes and branches (Fig 5). There are three types of nodes, root, internal and leaf. Leaf nodes correspond to the proteins in the tree. Root and internal nodes represent the inferred most common ancestor of the descendants. Branch length may be interpreted as time estimates between the nodes.

The root and internal nodes of the tree are shown as circles (speciation events) and squares (gene duplication events). If the tree has been previously curated, the nodes maybe colored in indicate the type of annotation (e.g., with inferred or experimental evidence). More details will be described in the "Making an inferrence" section of this guide. The nodes have unique identifiers that start with PTN (=PaNther Node), followed by 9 digits. Mouse over a node to see its identifier. If you right-click on a node, a menu will appear with the options to:
- Collapse node: - the entire clade is collapsed to a single node (rectangle). All the descendants are hidden, but the GO term assignments to them are still available for annotation. Right-click the node again and select "Expand node" to re-expand it.
- Reroot to node: - make the selected node and the root, and hide the rest of the tree. This is useful when the tree is too large. To bring back the entire tree again, use menu "Tree -> Reset Root to Main".
- Export seq ids from leaves: the ids of all leave sequences descended from the node are exported to a text file
- Prune: All nodes descended from the node are removed from the tree.
The tree branches can be rescaled if they are too long for comfortable viewing or too short to distinguish individual nodes. The default branch scale is 50, which works for most trees. To rescale, select Tree->Scale... and enter a different number.
- Click on a protein name in the tree to highlight the protein in the tree and the table.
- Left-click on a node in the tree to highlight the entire clade descended from it.
Annotation matrix
Note: The colors refer to the default colors in PAINT

The matrix has a row for each gene/gene product in the tree, and a column for each GO term that is directly annotated to at least one gene/gene product in the tree. The annotation matrix gives an overview of the annotations associated with any proteins in table format. It displays one of the three Gene Ontologies at a time. You can switch to a different ontology by clicking the radio button on the upper left part of the window (red arrow, Fig 6). Mouse-over the downward triangle to see the GO term (yellow circle). The terms in the annotation matrix are grouped, with the most specific terms on the left. A few very broad terms such as “protein binding” are not shown, even though they are listed in the Annotations pane.
Proteins with experimental annotations (IDA, EXP, IMP, IGI, IPI, or IEP evidence codes) for a particular ontology are colored and shown in boldface (blue circles). You may select one ontology at a time to examine using the radio buttons (red arrow) at the top of the window.
- Click on a protein in the tree and the corresponding row will be highlighted in the matrix.

- The annotations of the corresponding proteins and GO terms in the matrix are shown in colored squares (Fig 7).
- When you first open a tree, only the experimental annotations are shown. These are the annotations than can be used for annotating ancestral genes.
- Experimental annotations are represented by green color. If it is a direct annotation (i.e. the actual annotation is to that exact term in that column of the matrix), there is a black dot in the middle of the green square. If it is an indirect annotation (i.e. the actual annotation is to a child of the term in that column of the matrix), there is a white dot in the middle of the square.
- NOT annotations are indicated with by a red circle with a white X.
- When you have annotated an ancestral node, inferred annotations are also shown in the matrix. This allows you to easily keep track of what you've already annotated.
- Inferred annotations are represented by blue color, with either a black (direct) or white (indirect) dot in the center, or X for NOT as above.
- When you first open a tree, only the experimental annotations are shown. These are the annotations than can be used for annotating ancestral genes.
- Mouse-over an annotation square to see the tool tip of the protein name and the term.
- Click on the annotation square to highlight the row. All the annotations to the protein, as well as the evidences and confidence codes will be displayed in the Annotation panel (see below for more details).
- Right-click (or Command-click in Mac) on the experimental annotation (green square) in the matrix will automatically highlight the inferred most recent common ancestor (MRCA) node for the term.
Protein Information table

The phylogenetic tree is aligned with a protein information table showing additional information and linkouts to various databases (Fig 8). You can adjust the relative sizes of each within the window by dragging the line in the partition separating them. Note that the identifier table contains a lot of information that can be observed by scrolling to the right.
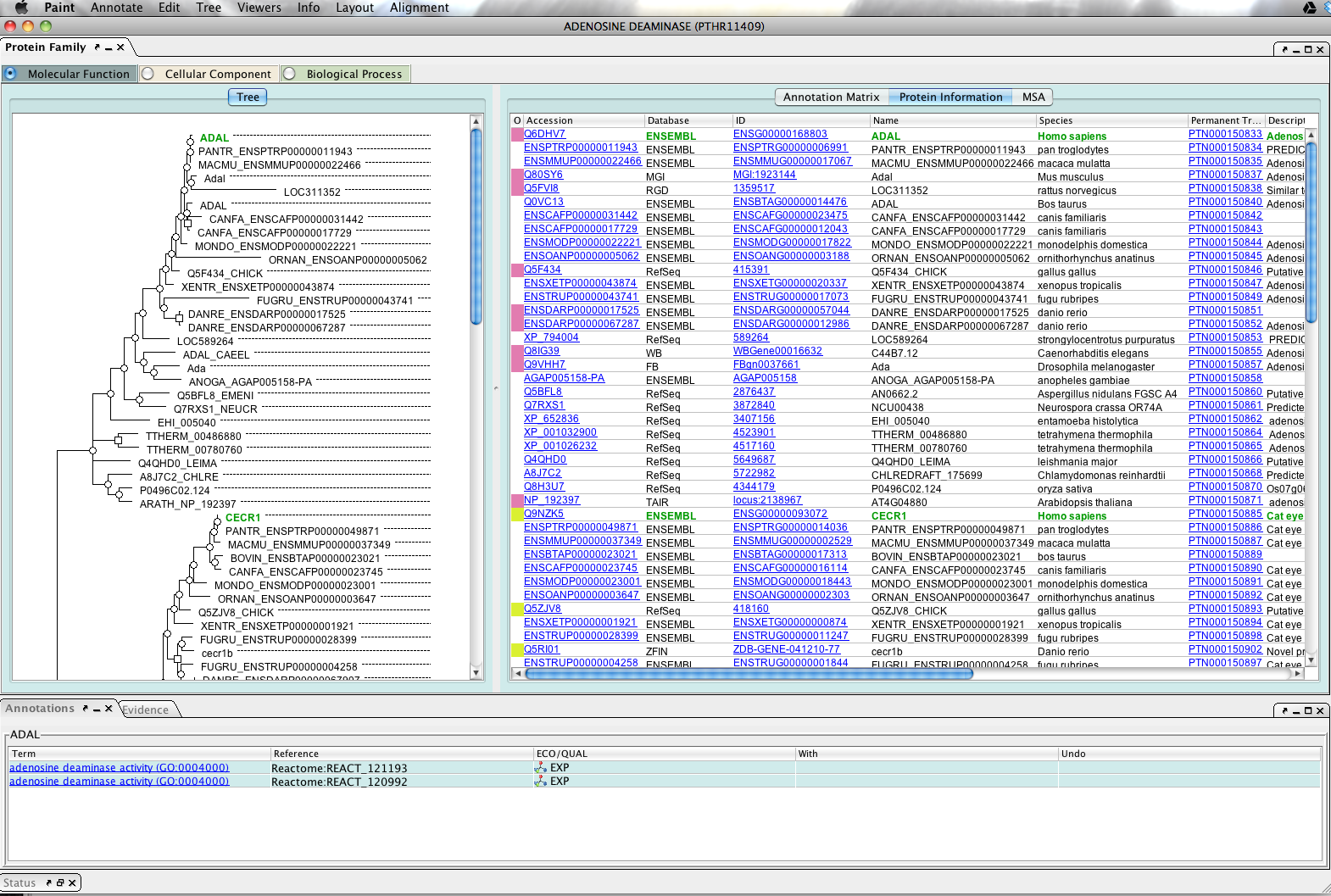{kind=link}
- Click anywhere within a row in the table to highlight the protein in the tree and the table.
- Click on one of the blue linkouts will open a link in your web browser.
Multiple sequence alignment (MSA)

The trees were estimated from an MSA (Figure 9). The evolutionarily conserved part of the alignment is indicated with uppercase letters. The other less conserved region is in lowercase letters. If a sequence misses a position in the matchstate, it is called a delete state and is designated by a dash. If a sequence needs to insert a position in the less conserved region in order to keep the match state region aligned, it is called an insert state and is designated by a dot.
{kind=link}
The conserved columns are colored with dark blue, blue or light blue, which indicates the conservation of 80%, 60% or 40%, respectively, in the column.
Toggle back and forth between the table view (“Protein Information”) and the MSA view (“MSA”) using the buttons above the table/MSA panel.
Note: You can view the sequence of a hypothetical ancestral protein (node) by first collapsing the appropriate node.
Annotations and Evidence windows


To view the annotations associated with a specific protein, click on that protein in the tree or table. Annotations appear in the Annotation pane (Fig 10), containing the following information:
- ECO (Evidence code): The type evidence code supporting the annotation.
- Term name: The GO term name and accession. Clicking on the term links out to AmiGO. A term with a NOT annotation is displayed as strikethrough text.
- Reference: The reference supporting the annotation. Clicking on the reference links out to PubMed. IBA annotations display an internal reference, PAINT_REF:00nnnnn, where nnnnn is the numerical part of the Panther family ID.
- With: This column contains the evidence to support the inference.
- Qualifiers: The qualifiers 'NOT', 'colocalizes_with' and 'contributes_to' each have a column in the annotation table, and a checkbox that is checked when the qualifier is present.
- Delete: This is used to remove an inference made by PAINT (see Removing an IBD annotation section below).
Evidence window
The evidence window displayed automatically generated logs of the tree curation, such as annotations, validation and changes made by the PAINT pipeline upon data release.
Curator notes
The Curator notes is a text editor used to record notes on the curation process. Curator notes can be modified by going to the File > Update comment menu.
NOTE: The purpose of the annotation notes is to convey important points about the annotations and the phylogenetic tree both to other annotators and to users, so annotators should try to make the notes as clear as possible.
The annotator may use the Curator notes to describe important points in the annotation process, including:
- References used to annotate the family (for example, a few major reviews)
- Any important points about the family topology, including potential inconsistencies in the tree
- Reasons for annotating to a different node than the MRCA (most common recent ancestor), ie the node that triangulation of annotation identifies.
- Link to GitHub tickets leading to review of the tree annotation.
“Find” function

The Find function (Edit > Find…, Fig 12A) allows you to search for either a gene or a GO term. Select a gene or term search using the radio buttons (Fig 12B). Searches are case-insensitive.
A gene search matches against exact match of any text stored in the database, such as any sequence identifiers, gene symbol, or even gene name (red arrow, Fig 12C). The search does not return partial match (blue arrow, (Fig 12C). To do a partial match, wildcard character(s) (*) can be added before and/or after the search term. Scroll through the list of matches and click on a specific match to highlight it in the tree, table, and annotation matrix, and to display its annotations in the Annotations window.
You may search GO terms using text, or you may use numbers to search for GO IDs.
Making an inference: Transferring annotations
Ancestral nodes in the tree can be annotated with any GO term that has been annotated to one (or more) of its descendants. These “inferred” annotations can be propagated to its other descendants.
Annotating an ancestral node, and propagating to descendants by inheritance

- In the example shown in Fig 13A, 5 proteins are annotated by EXP to the GO term
adenine deaminase activity(green squares in the 1st column of the Annotation Matrix, indicated by the red downward arrow).
Tip: To view the last common ancestor that can be annotated based on triangulation(*), right click on a GO term, or anywhere in the column that contains that GO term. An inferred node, as well as its descendants, will be highlighted in grey (blue arrow, Fig 13A).
- (*) Triangulation is the calculation of the last common ancestor of two sequences; in this case PAINT calculates the last common ancestor supporting an annotation to the same GO term.
- Note that the node calculated by triangulation may not be the best on to annotate: the curator can decide to annotate to an more ancient or a more recent ancestor, dependent on other evidence. For example, annotations in other GO aspects may support an earlier annotation than suggested by the triangulation.
- The curator should not assume that the genes in the tree are fully curated with repsect to the primary literature. It may be useful to review other papers, including reviews, to ensure annotations found in the tree accurately represents the current state of knowledge.
To annotate an ancestral node:

- Click a GO term (green square) from the Annotation Matrix (Fig. 13B) (or anywhere in the column containing the GO term).
- Drag the term to the ancestral node you wish to annotate. This can be the inferred node or any other nodes. When you mouse over it, a
+ signwill be visible next to the node. Release the mouse button to annotate. Click here for a video demo of the procedure: http://youtu.be/8kHrdiuNfos. - The node is now annotated with that term using the evidence code “IBD” (“Inferred from Biological Descendant”).
- PAINT then automatically propagates the IBD annotation to all descendants of the PAINTEed node, such that all descendants of the node will now be annotated with that term using the evidence code “IBA” (“Inferred from Biological Ancestor”) (Fig. 13B). (Proteins and nodes already annotated with the term or one of its descendant terms will remain unchanged.)
Notes:
- If you propagate an experimental annotation that has a qualifier, e.g. "NOT", "contributes_to" (for MF annotations), or "colocalizes_with" (for CC annotations), the qualifier will also get propagated, unless there are contradictory annotations, ie, annotations with and annotation without the qualifier (for the same or for different genes). In that case, a pop-up window will appear to specify whether the qualifier(s) should be propagated. The default is No; to accept this option, click OK. To propagate the qualifier, tick the Yes button and click OK.
- You may only annotate a node with a given GO term if AT LEAST ONE descendant has an annotation to that term or a child term. If a node does not turn dark (step 2), it cannot be annotated.
- Effectively, the PAINT curator only makes IBD annotations; IBA annotations are generated automatically to all descendants of the node to which an IBD annotation is made.
- The IBD annotation automatically includes evidence for each of the sequences having an EXP annotation to the term or one if its descendants; it is not necessary to propagate individual EXP.
Removing an IBD annotation
IBD annotations can be removed as followed:
- Click on the desired node. Nodes with inferred annotations are colored orange (Fig. 13B).
- Go to the Annotation tab. To remove an annotation, click the
Deletein the Delete column (in a red square in Fig. 13B).
Negation of annotations: "NOT" statements
Background: Since PAINT is a model of the family's evolution, adding a NOT modifier to a descendant (either another node or a leaf) represents a loss of function during evolution, that is, we are stating that the specified function was present in an ancestral protein and has been lost in the indicated protein or clade. This is a special case of the GO guidelines for NOT, which state that a NOT annotation may be made in situations where a particular function may be expected but is absent.
PAINT defines two reasons for an evolutionary loss of function, described with two separate evidence codes (ECO):
- IRD = Inferred from Rapid Divergence from ancestral sequence evidence used in manual assertion: Used when there is a long branch, often following a duplication, and significant sequence divergence. For very divergent sequences, predictions are less reliable, even in the presence of a common ancestor.
- IKR = Inferred from phylogenetic determination of loss of Key Residues evidence: Used when a residue known to be required for the activity of the protein has mutated.
In both cases, the node (intermediate or leaf) on which the NOT annotation is placed gets the evidence code selected (IKR or IRD), and descendants, if any, are annotated with the IBA evidence.
You may add a NOT modifier to an IBA annotation if the evidence warrants it, listed here in order of decreasing strength:
- There are experimental annotations indicating that a function has been lost from one or more proteins.
- Specific residues have been mutated at, for example, an enzyme’s catalytic site, and a specified function is no longer possible. (Evidence code IKR = Inferred from Known Residues)
- A protein or clade has evolved rapidly, losing the original function and gaining a new one. This may be visible as a long branch in the tree, but the meaning of “long” varies by context, and a visibly long branch is not strictly required. (Evidence code IRD = Inferred from Rapid Divergence)
To add the NOT qualifier:
- Select a node or protein from the tree. This may be either a directly annotated node or one of its children.
- In the ECO/QUAL column of the Associations window, click on the “IAS.” A popup menu will appear.
- Under “NOT,” select which evidence code justifies the NOT qualifier.
- If the evidence code is IBA or IKR, all annotations to proteins and nodes descended from that node will have the NOT qualifier added (as these have good evidence for loss)
- If the evidence code is IRD, descendant sequences will not be annotated with the NOT qualifier, but the ancestral annotation will not be propagated to the descendants. Thus this acts like a STOP PROPAGATION.
You may remove a NOT qualifier by clicking the Delete box in the Delete column. Note that you can only remove the qualifiers from the specific node to which it was made.
In addition to the annotation no longer propagating downward, a small hash mark will appear near the node in the tree to indicate that the block exists. Note that a hash mark only indicates the existence of at least one block, not that every annotation through that node is blocked.
Partial annotation of trees
 |
When you want to annotate a very large family, e.g. the RAB GTPase superfamily (PTHR24073), it may not be feasible to annotate all clades at the same time. In this kind of situation, you may choose to annotate only the clades you are knowledgeable and confident of, and leave other clades unexamined. When you do this, you should fully annotate the clades you choose to annotate. For example, if you choose to do the IFT27 clade, do it fully. Please don't do piecemeal annotations in various locations that may make it hard for a subsequent annotator to understand what has been done.
We also agreed at the July 2014 PAINT Jamboree that you can make propagations all the way to the root if you feel that there is an ancestral role, even if you think that some clades have lost this. For example, in the RAB GTPase superfamily, we think that it had an ancestral function as a GTPase, but it is possible that some clades, e.g. the IFT22 clade, have lost this ancestral activity. You can make these high level propagations as part of your initial annotation of the family. If there are clades where this is wrong, perhaps the IBA annotation from PAINT will generate feedback that will help us correct it.
Recording partial annotation in the notes file
If you only partially annotate a tree, please record in the notes file which clades you have worked on using the node number, e.g. Eukaryota_PTN001180007 as well as a common name, e.g. IFT27, if it is helpful.
Recording trees examined, but not annotated
When you examine a tree and feel that it should not be annotated for some reason, please record that in the Evidence Notes so that we can track the fact that the family has been examined. Please use one of these tags (in all caps) in the Notes section of the Evidence tab. You can additional information after the tag if you wish (syntax between tag and additional info not discussed or determined). Then, save your annotations as normal so that PAINT will save the notes file.
- MISSING ANNOTATION - Use this if the tree looks OK, but there are insufficient experimental annotations to propagate any annotations.
- MISSING SEQUENCE - Use this if you feel that a specific sequence or sequences is missing. You can list the IDs of the sequence(s) after the tag.
- BAD TREE - Use this if you feel that the tree has major problems beyond one or a few missing sequences.
Interpreting the PANTHER trees
Speciation and duplication events, and horizontal transfer
In the tree, a speciation node is shown with a circle, and a gene duplication node with an square. Horizontal transfer events also appear in the tree, though more rarely, and these are represented with a diamond.
Branch lengths
- Branch lengths show the amount of sequence divergence that has occurred between a given node and its ancestral node, in terms of the average number of amino acid substitutions per site. Shorter branches indicate less sequence divergence and therefore greater conservation of ancestral characters. A branch might be shorter because of a slower evolutionary rate (greater negative selection), or because less "time" has gone by (actually a combination of number of generations and population dynamics), or both.
- Very long branches indicate an unreliable divergence estimate, due to insufficient data. Note that sometimes there is not enough data to compare all branches that descend from a given node. In this case, we have set all descendant branches to a length of 2.0 (very long branches). Branch lengths of 2.0 are often due to a sequence fragment, and at a duplication node it may also indicate a gene that has been incorrectly broken into two different genes by a gene prediction program.
- Following a gene duplication (after a square node), the relative branch lengths for descendant branches are particularly useful: the shortest branch (least diverged) is more likely to have greater functional conservation.
Multiple sequence alignment (MSA)
- Some columns in the MSA have upper-case characters (and dashes '-' for insertions/deletions). These columns were used to estimate the phylogenetic tree.
- Lower-case characters and periods (‘.’ for insertions/deletions) denote positions that were ignored when estimating the phylogenetic tree. Sometimes, tree errors arise because not enough columns were used, and the phylogeny could not be reconstructed well based on the included columns. Since they were not used in the phylogeny, lower-case characters can be particularly helpful in verifying the tree topology: any conserved insertions should be parsimoniously traceable to a common ancestor.
Reporting bugs or likely errors in the trees
Tree issues
Most often, the errors in phylogenetic trees are due to problems with the sequence alignment, or the specific MSA columns used to estimate the phylogeny. The phylogeny inference program performs fairly robust handling of sequence fragments, but sequence fragments still cause errors. Another source of error is when the sequences evolve very slowly, generating little variation from which to estimate phylogeny. In this case, the errors can usually be fixed by including additional alignment positions to consider in the phylogeny.
If a Panther tree needs to be reviewed, please create a ticket in the Panther GitHub tracker: https://github.com/pantherdb/Helpdesk/issues
PAINT issues
Issues with the PAINT tools should be reported in this tracker: https://github.com/pantherdb/db-PAINT/issues
Curation Guidelines
Those guidelines have been published (Gaudet, Livestone, Lewis, Thomas, 2011) [1]
Curation guidelines are described in detail on this page: http://wiki.geneontology.org/index.php/PAINT_SOP